
For most people, breathing is something you don’t think about. But for someone with cystic fibrosis (CF), every breath can feel like a battle. Thick, sticky mucus clogs the lungs, traps bacteria, and leads to constant infections. It’s not just the lungs-this disease affects the pancreas, liver, and reproductive system too. What makes CF different from other respiratory illnesses is that it’s not caused by smoking, pollution, or viruses. It’s genetic. And for decades, there was little doctors could do but manage the symptoms.
What Exactly Is Cystic Fibrosis?
Cystic fibrosis is a life-limiting inherited condition caused by mutations in the CFTR gene. This gene makes a protein that acts like a gatekeeper for salt and water in and out of cells. When it’s broken, the body produces mucus that’s thick and sticky instead of thin and slippery. That mucus builds up in the lungs, pancreas, and other organs, causing serious problems.
The disease only shows up if a person inherits two bad copies of the gene-one from each parent. If you have just one, you’re a carrier. You won’t have symptoms, but you can pass the mutation to your kids. About 1 in 25 people of Northern European descent carry a CF mutation. That’s why it’s more common in white populations, though it affects people of all backgrounds.
Doctors have known about CF since the 1930s, but it wasn’t until 1989 that the exact gene was found. Since then, we’ve learned there are over 2,000 different mutations in the CFTR gene. The most common one, called F508del, shows up in about 70% of cases worldwide. Other mutations are rarer, and that’s where things get complicated-because not all treatments work for all mutations.
How CF Affects the Body
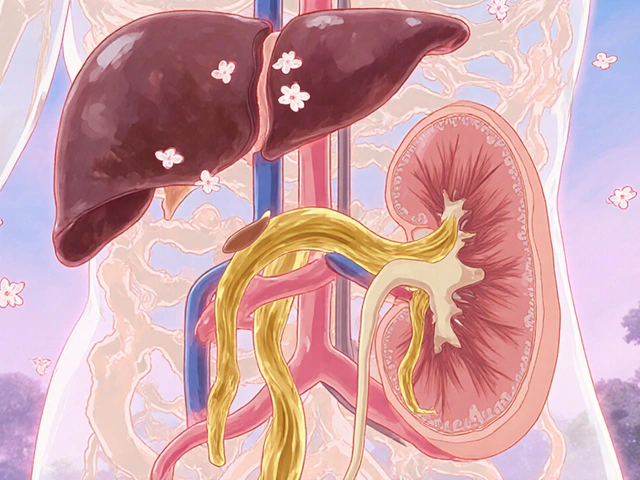
CF doesn’t just mess with breathing. The thick mucus clogs the tiny tubes in the pancreas, stopping digestive enzymes from reaching the intestines. About 85% of people with CF need to take enzyme pills with every meal just to absorb nutrients. Without them, they can’t gain weight or grow properly-even if they eat a lot.
In the lungs, mucus becomes a breeding ground for bacteria. Pseudomonas aeruginosa and Staphylococcus aureus are the usual suspects. These infections lead to inflammation, scarring, and permanent lung damage called bronchiectasis. Over time, lung function drops. That’s why respiratory failure is the leading cause of death in CF-responsible for about 85% of fatalities.
Other organs aren’t spared. About 30% of people develop liver problems from blocked bile ducts. And nearly all men with CF are infertile because they’re born without the tube that carries sperm. Women can have children, but pregnancy comes with higher risks.
One of the easiest ways to diagnose CF is the sweat test. People with CF have unusually salty sweat-chloride levels above 60 mmol/L. That’s why newborn screening now includes this test in all 50 U.S. states. Early diagnosis means early treatment, which makes a huge difference.
The Old Way: Managing Symptoms
Before 2012, treatment was all about damage control. People with CF spent hours every day doing airway clearance-hitting their chest with a vest, shaking their lungs with a handheld device, or doing breathing exercises. They inhaled up to six different medications daily: antibiotics to fight infections, bronchodilators to open airways, and mucus-thinning drugs.
They also took up to 12 pancreatic enzymes per meal, vitamin supplements, and high-calorie shakes just to stay alive. The daily routine could take 2 to 3 hours. For kids, it meant missing school activities. For adults, it meant missing work or social events.
Adherence was a huge problem. Studies show only 65-75% of people stuck with the full regimen. It’s exhausting. And even with all that effort, life expectancy was grim. In 1960, the median survival was just 14 years. By 2000, it had crept up to 30. But then everything changed.

The Game Changer: CFTR Modulators
The first real breakthrough came in 2012 with the approval of ivacaftor (Kalydeco). It was the first drug that didn’t just treat symptoms-it fixed the broken CFTR protein itself. It worked for people with the G551D mutation, which affects about 4% of CF patients. In clinical trials, users saw a 10.6% jump in lung function. That’s huge. For comparison, most asthma inhalers improve lung function by 1-3%.
Then came the real revolution: Trikafta (elexacaftor/tezacaftor/ivacaftor), approved in 2019. This triple combo works for about 90% of people with CF, including those with two copies of F508del-the most common mutation. In trials, Trikafta boosted lung function by 13.8% and cut lung flare-ups by 63%. That’s not just an improvement-it’s a transformation.
One 28-year-old patient on a CF forum said their daily airway clearance dropped from 90 minutes to 20 minutes after starting Trikafta. They went from being too tired to leave the house to hiking on weekends. That’s the kind of change that doesn’t show up in lab reports-it shows up in real life.
By 2023, 90% of people with CF in the U.S. had access to at least one modulator. Life expectancy jumped to 50.9 years. For the first time, most people with CF are adults. In 1990, only 27% were over 18. Now it’s 52%.
The Dark Side of Progress
But this miracle comes with a price tag. Trikafta costs about $300,000 a year in the U.S. Even with insurance, many families pay $1,200 a month out of pocket. A 2022 survey found 42% of modulator users felt financial strain. That’s why only 35% of people with CF worldwide have access to these drugs. In low-income countries, the number is under 10%.
There are side effects too. About 3.2% of users develop liver enzyme spikes so severe they have to stop the drug. Some report headaches, rashes, or cataracts. And not everyone responds. About 10% of patients have mutations that current modulators can’t fix-mostly those with Class I mutations, where the protein doesn’t get made at all.
One case from the European registry tells the story: a 34-year-old man with a rare mutation kept getting worse despite aggressive therapy. He had no modulator option. His lungs kept deteriorating. He didn’t die from infection-he died because there was no drug that could help him.
What’s Next? The Fight for the Remaining 10%
The Cystic Fibrosis Foundation has poured over $750 million into research since 1989. That investment didn’t just fund science-it changed how drug development works. Their venture philanthropy model gave $150 million to a small biotech firm in 2000. That company became Vertex Pharmaceuticals, which now controls 95% of the CF drug market.
Today, 15 new therapies are in clinical trials. Some aim to fix nonsense mutations using mRNA technology. Others use CRISPR gene editing to repair the faulty gene directly. There are even new inhaled antibiotics designed to kill stubborn Pseudomonas strains that survive even the strongest drugs.
The Foundation’s new $100 million "Path to a Cure" initiative is focused entirely on the 10% left behind. They’re not waiting for big pharma to act. They’re funding the science themselves.
Life With CF Today
If you were diagnosed with CF in 1980, your life expectancy was 25. If you’re diagnosed today, it’s over 50. That’s not just medicine-it’s a cultural shift. CF care centers are now specialized hubs with teams of doctors, dietitians, physical therapists, and social workers. There are 260 accredited centers in the U.S. alone.
Support networks are stronger than ever. Online communities like CF Buddy Connect have over 12,500 active users. The annual International CF Conference draws thousands of patients, families, and researchers. People are talking openly about mental health, fertility, employment, and insurance battles.
But the biggest change isn’t in the drugs. It’s in the mindset. CF is no longer a death sentence. It’s a chronic condition-with a future. The goal now isn’t just to survive. It’s to live well.
What You Need to Know
- CF is genetic, not contagious. Two bad copies of the CFTR gene are needed.
- Over 2,000 mutations exist. F508del is the most common (70%).
- CFTR modulators like Trikafta treat the root cause-not just symptoms.
- 90% of U.S. patients now have access to modulators. Globally, it’s 35%.
- Life expectancy has jumped from 14 years in 1960 to 50.9 years in 2022.
- 10% of patients still have no effective modulator therapy.
- Cost remains the biggest barrier to global access.
Is cystic fibrosis curable?
No, cystic fibrosis is not yet curable. But CFTR modulator therapies like Trikafta have turned it from a fatal childhood disease into a manageable chronic condition for most people. Scientists are working on gene editing and mRNA treatments that could one day fix the faulty gene entirely, but those are still in testing.
Can you get cystic fibrosis if only one parent is a carrier?
No. CF is an autosomal recessive disorder, meaning you must inherit two mutated copies of the CFTR gene-one from each parent. If only one parent is a carrier, the child might be a carrier too, but they won’t have the disease. Both parents must be carriers for a child to have a 25% chance of developing CF.
How do CFTR modulators work?
CFTR modulators are drugs that fix the broken CFTR protein. Some help the protein fold correctly (correctors like tezacaftor), others help it open to let salt through (potentiators like ivacaftor). Trikafta combines both types to restore function in the most common mutations. They don’t change your DNA-they just help the protein do its job.
Why are CFTR modulators so expensive?
They’re expensive because they’re targeted therapies developed for a small group of patients (orphan drugs). The research took decades and billions in funding. Vertex Pharmaceuticals, the main maker, holds patents and market exclusivity. In the U.S., insurance often covers them, but out-of-pocket costs can still be high. In many countries, they’re simply not available due to cost.
Are there side effects to CFTR modulators?
Yes. Common side effects include headaches, nausea, and elevated liver enzymes. In about 3.2% of users, liver damage is severe enough to require stopping the drug. Some people develop cataracts or skin rashes. Long-term effects are still being studied, but for most, the benefits far outweigh the risks.
Can someone with CF have children?
Most men with CF are infertile due to missing or blocked tubes that carry sperm, but they can still father children using assisted reproductive techniques. Women with CF can get pregnant, but pregnancy carries higher risks for both mother and baby due to lung function decline and nutritional demands. Fertility counseling is now standard care.
What’s the biggest challenge facing CF patients today?
Access. While 90% of patients in the U.S. have access to life-changing modulators, only 35% globally do. In low-income countries, many people still die from complications that could have been prevented. Cost, lack of infrastructure, and limited screening are the main barriers. Equity in treatment is now the biggest unmet need in CF care.
CF used to be a disease of hospitals and hospice. Now it’s a disease of clinics, coffee shops, and college campuses. People with CF are graduating, working, traveling, and raising families. The science behind their survival is remarkable. But the real victory isn’t in the lab-it’s in the everyday moments they now get to live.




12 Comments
Hannah Magera
November 29, 2025 AT 03:10I never realized how much CF care has changed until I saw a friend’s kid start Trikafta. The difference in energy is wild-suddenly they’re running around like any other kid. I used to think it was all about meds and machines, but it’s really about giving people back their time. No more 3-hour routines. No more missing school. Just... breathing easy.
It’s not perfect, but it’s a start. And honestly? That’s more than we had 10 years ago.
Austin Simko
November 30, 2025 AT 11:30Trikafta was funded by the government. They just let Vertex take the credit.
Nicola Mari
December 1, 2025 AT 12:44It’s heartbreaking to see how easily we celebrate pharmaceutical miracles while ignoring the people left behind. The 10% who can’t afford it, the countries without screening, the families who still bury their children. This isn’t progress-it’s a luxury for the wealthy. And we call it medicine?
Sam txf
December 2, 2025 AT 12:31Vertex is a monopoly with a monopoly on hope. They didn’t ‘invent’ CF treatment-they bought every competitor, locked down patents, and turned a genetic disease into a cash cow. And now they’re smugly patting themselves on the back while families in India are selling their land to buy a month’s supply. This isn’t science. It’s capitalism with a white coat.
George Hook
December 4, 2025 AT 05:20I’ve been following CF research since the late 90s, and honestly, the shift from symptom management to targeted protein correction is one of the most profound advances in modern medicine. It’s not just about lung function numbers-it’s about identity. People with CF are no longer defined by their illness. They’re students, artists, athletes, parents. The modulators didn’t just extend life-they restored personhood.
But I agree with Nicola: the equity gap is terrifying. We’ve created a two-tier system where survival depends on zip code and insurance plan. That’s not a medical failure-it’s a moral one. And until we fix that, no amount of science should make us feel proud.
jaya sreeraagam
December 4, 2025 AT 16:19As someone from India, I want to say this: I’ve seen families in rural areas who don’t even know what CF is. No newborn screening, no access to sweat tests, no modulators-nothing. The fact that life expectancy jumped to 50 in the US is incredible, but here, many kids still die before age 5 from pneumonia that could’ve been managed. We need global funding, not just fancy drugs for rich countries. The CF Foundation’s $100M initiative? Brilliant-but it needs to be matched by WHO and UNICEF. This isn’t just a US problem. It’s a human problem.
Katrina Sofiya
December 5, 2025 AT 12:24Every single person who has lived with CF and found a way to thrive deserves to be celebrated. The courage it takes to wake up every day and fight-even before Trikafta-is unimaginable. And now, with these therapies, we’re witnessing a renaissance. People are going to college, falling in love, starting businesses. The future isn’t just longer-it’s brighter. Let’s keep pushing for equity, yes, but let’s also honor the resilience that got us this far. You are not your disease. You are your strength.
kaushik dutta
December 5, 2025 AT 15:43The biotech model is fundamentally broken. The CF Foundation’s venture philanthropy was genius, sure-but it created a monoculture. Vertex now controls 95% of the market. No competition. No price pressure. No incentive to develop drugs for rare mutations because the ROI is too low. We need public R&D, not corporate charity. CRISPR and mRNA should be open-source, not patented. The science belongs to humanity, not shareholders. And until we dismantle this profit-driven paradigm, the 10% will keep dying in silence.
doug schlenker
December 5, 2025 AT 22:26I just want to say thank you to everyone who’s lived with CF and kept going-even when the treatments were brutal. I’ve met parents who did chest physio for hours every day while working two jobs. I’ve talked to teens who hid their nebulizers in their backpacks so they wouldn’t get teased. This isn’t just about drugs. It’s about dignity. And Trikafta didn’t just improve lung function-it gave people back their right to be ordinary. That’s worth more than any statistic.
Olivia Gracelynn Starsmith
December 7, 2025 AT 08:49The sweat test is still the gold standard and it’s so simple. I wish more people knew that. And Trikafta? It’s not a cure but it’s the closest thing we’ve had. The drop in hospital visits alone is staggering. But yeah, the cost is insane. I work in public health and we’re seeing families choose between rent and meds. We need policy change, not just hope.
Skye Hamilton
December 7, 2025 AT 10:19Everyone’s acting like Trikafta is a miracle but what about the people who got worse on it? My cousin developed liver failure after six months. They told her it was ‘rare’ but it happened to her. And now she’s on transplant list. So don’t act like this is all sunshine. The system is rigged and the side effects are buried in fine print. Also, why is it called a ‘cure’ in the headlines when it’s not? Clickbait.
Maria Romina Aguilar
December 7, 2025 AT 15:22Wait-so… the gene therapy is still experimental? But the drug costs $300,000? And you’re saying it’s not a cure? So… we’re paying a fortune for a treatment that doesn’t fix the root cause, and only works for 90%? And the other 10%? They’re just… out of luck? And we’re supposed to be grateful? This is terrifying.